Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors.
ATC code: L01XE05.
Pharmacology: Pharmacodynamics: Sorafenib is a multikinase inhibitor which has demonstrated both anti-proliferative and anti-angiogenic properties in vitro and in vivo.
Mechanism of action and pharmacodynamic effects: Sorafenib is a multikinase inhibitor that decreases tumour cell proliferation in vitro. Sorafenib inhibits tumour growth of abroad spectrum of human tumour xenografts in athymic mice accompanied by a reduction of tumour angiogenesis.
Sorafenib inhibits the activity of targets present in the tumour cell (CRAF, BRAF, V600E BRAF, c-KIT, and FLT-3) and in the tumour vasculature (CRAF, VEGFR-2, VEGFR-3, and PDGFR-β). RAF kinases areserine/threonine kinases, whereas c-KIT, FLT-3, VEGFR-2, VEGFR-3, and PDGFR-β are receptor tyrosine kinases.
Clinical efficacy: The clinical safety and efficacy of sorafenib have been studied in patients with hepatocellular carcinoma (HCC), in patients with advanced renal cell carcinoma (RCC) and in patients with differentiated thyroid carcinoma (DTC).
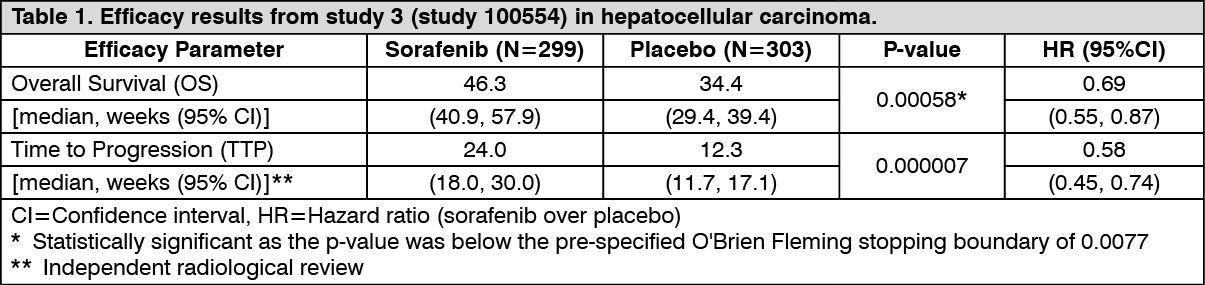
Hepatocellular carcinoma: Study 3 (study 100554) was a Phase III, international, multi-centre, randomised, double blind, placebo-controlled study in 602 patients with hepatocellular carcinoma. Demographics and baseline disease characteristics were comparable between the sorafenib and the placebo group with regard to ECOG status (status 0: 54% vs. 54%; status 1: 38% vs. 39%; status 2: 8% vs. 7%), TNM stage (stage I: <1% vs. <1%; stage II: 10.4% vs. 8.3%; stage III: 37.8% vs. 43.6%; stage IV: 50.8% vs. 46.9%), and BCLC stage (stage B: 18.1% vs. 16.8%; stage C: 81.6% vs. 83.2%; stage D: <1% vs. 0%).
The study was stopped after a planned interim analysis of OS had crossed the pre-specified efficacy boundary. This OS analysis showed a statistically significant advantage for sorafenib over placebo for OS (HR: 0.69, p=0.00058, see table 1).
There are limited data from this study in patients with Child Pugh B liver impairment and only one patient with Child Pugh C had been included. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A second Phase III, international, multi-centre, randomised, double blind, placebo-controlled study (Study 4, 11849) evaluated the clinical benefit of sorafenib in 226 patients with advanced hepatocellular carcinoma. This study, conducted in China, Korea and Taiwan confirmed the findings of Study 3 with respect to the favourable benefit-risk profile of sorafenib (HR (OS): 0.68, p=0.01414).
In the pre-specified stratification factors (ECOG status, presence or absence of macroscopic vascular invasion and/or extra-hepatic tumour spread) of both Study 3 and 4, the HR consistently favoured sorafenib over placebo. Exploratory subgroup analyses suggested that patients with distant metastases at baseline derived a less pronounced treatment effect.
Renal cell carcinoma: The safety and efficacy of sorafenib in the treatment of advanced renal cell carcinoma (RCC) were investigated in two clinical studies: Study 1 (study 11213) was a Phase III, multi-centre, randomised, double blind, placebo-controlled study in 903 patients.
Only patients with clear cell renal carcinoma and low and intermediate risk MSKCC (Memorial Sloan Kettering Cancer Center) were included. The primary endpoints were overall survival and progression-free survival (PFS).
Approximately half of the patients had an ECOG performance status of 0, and half of the patients were in the low risk MSKCC prognostic group.
PFS was evaluated by blinded independent radiological review using RECIST criteria. The PFS analysis was conducted at 342 events in 769 patients. The median PFS was 167 days for patients randomised to sorafenib compared to 84 days for placebo patients (HR=0.44; 95% CI: 0.35-0.55; p <0.000001). Age, MSKCC prognostic group, ECOG PS and prior therapy did not affect the treatment effect size.
An interim analysis (second interim analysis) for overall survival was conducted at 367 deaths in 903 patients. The nominal alpha value for this analysis was 0.0094. The median survival was 19.3 months for patients randomised to sorafenib compared to 15.9 months for placebo patients (HR=0.77; 95% CI: 0.63-0.95; p=0.015). At the time of this analysis, about 200 patients had crossed-over to sorafenib from the placebo group.
Study 2 was a Phase II, discontinuation study in patients with metastatic malignancies, including RCC. Patients with stable disease on therapy with sorafenib were randomised to placebo or continued sorafenib therapy. Progression-free survival in patients with RCC was significantly longer in the sorafenib group (163 days) than in the placebo group (41 days) (p=0.0001, HR=0.29).
Differentiated thyroid carcinoma (DTC): Study 5 (study 14295) was a Phase III, international, multi-centre, randomised, double blind, placebo-controlled trial in 417 patients with locally advanced or metastatic DTC refractory to radioactive iodine. Progression-free survival (PFS) as evaluated by a blinded independent radiological review using RECIST criteria was the primary endpoint of the study.
Secondary endpoints included overall survival (OS), tumour response rate and duration of response. Following progression, patients were allowed to receive open label sorafenib.
Patients were included in the study if they experienced progression within 14 months of enrollment and had DTC refractory to radioactive iodine (RAI). DTC refractory to RAI was defined as having a lesion without iodine uptake on a RAI scan, or receiving cumulative RAI ≥22.2 GBq, or experiencing a progression after a RAI treatment within 16 months of enrollment or after two RAI treatments within 16 months of each other.
Baseline demographics and patient characteristics were well balanced for both treatment groups. Metastases were present in the lungs in 86%, lymph node in 51% and bone in 27% of the patients. The median delivered cumulative radioactive iodine activity before enrollment was approximately 14.8 GBq. Majority of patients had papillary carcinoma (56.8%), followed by follicular (25.4%) and poorly differentiated carcinoma (9.6%).
Median PFS time was 10.8 months in the sorafenib group compared to 5.8 months in the placebo group (HR=0.587; 95% Confidence Interval (CI): 0.454, 0.758; one-sided p <0.0001).
The effect of sorafenib on PFS was consistent independent of geographic region, age above or below 60 years, gender, histological subtype, and presence or absence of bone metastasis.
In an overall survival analysis conducted 9 months after the data cut-off for the final PFS analysis there was no statically significant difference in overall survival between the treatment groups (HR=0.884; 95% CI: 0.633, 1.236, one-sided p value of 0.236). The median OS was not reached in the sorafenib arm and was 36.5 months in the placebo arm. One hundred fifty seven (75%) patients randomised to placebo and 61 (30%) patients randomised to sorafenib received open-label sorafenib.
The median duration of therapy in the double-blind period was 46 weeks (range 0.3-135) for patients receiving sorafenib and 28 weeks (range 1.7-132) for patients receiving placebo.
No complete response (CR) according to RECIST was observed. The overall response rate (CR + partial response (PR) per independent radiological assessment was higher in the sorafenib group (24 patients, 12.2%) than in the placebo group (1 patient, 0.5%), one-sided p<0.0001. The median duration of response was 309 days (95% CI: 226, 505 days) in sorafenib treated patients who experienced a PR.
A post-hoc subgroup analysis by maximum tumour size showed a treatment effect for PFS in favour of sorafenib over placebo for patients with maximum tumour size of 1.5 cm or larger (HR 0.54 (95% CI: 0.41-0.71)) whereas a numerically lower effect was reported in patients with a maximum tumour size of less than 1.5 cm (HR 0.87 (95% CI: 0.40-1.89).
A post-hoc subgroup analysis by thyroid carcinoma symptoms at baseline showed a treatment effect for PFS in favour of sorafenib over placebo for both symptomatic and asymptomatic patients. The HR of progression free survival was 0.39 (95% CI: 0.21-0.72) for patients with symptoms at baseline and 0.60 (95% CI: 0.45-0.81) for patients without symptoms at baseline.
QT interval prolongation: In a clinical pharmacology study, QT/QTc measurements were recorded in 31 patients at baseline (pre-treatment) and post-treatment. After one 28-day treatment cycle, at the time of maximum concentration of sorafenib, QTcB was prolonged by 4 ±19 msec and QTcF by 9±18 msec, as compared to placebo treatment at baseline. No subject showed aQTcB or QTcF >500 msec during the post-treatment ECG monitoring.
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies, in all subsets of the paediatric population, in kidney and renal pelvis carcinoma (excluding nephroblastoma, nephroblastomatosis, clear cell sarcoma, mesoblasticnephroma, renal medullary carcinoma and rhabdoid tumour of the kidney) and liver and intrahepatic bile duct carcinoma (excluding hepatoblastoma) and differentiated thyroid carcinoma.
Pharmacokinetics: Absorption and distribution: After administration of sorafenib tablets the mean relative bioavailability is 38-49% when compared to an oral solution.
The absolute bioavailability is not known. Following oral administration sorafenib reaches peak plasma concentrations in approximately 3 hours. When given with a high-fat meal sorafenib absorption was reduced by 30% compared to administration in the fasted state.
Mean Cmax and AUC increased less than proportionally beyond doses of 400 mg administered twice daily. In vitro binding of sorafenib to human plasma proteins is 99.5%.
Multiple dosing of sorafenib for 7 days resulted in a 2.5- to 7-fold accumulation compared to single dose administration.
Steady state plasma sorafenib concentrations are achieved within 7 days, with a peak to trough ratio of mean concentrations of less than 2.
The steady-state concentrations of sorafenib administered at 400 mg twice daily were evaluated in DTC, RCC and HCC patients. The highest mean concentration was observed in DTC patients (approximately twice that observed in patients with RCC and HCC), though variability was high for all tumour types. The reason for the increased concentration in DTC patients is unknown.
Biotransformation and elimination: The elimination half-life of sorafenib is approximately 25-48 hours. Sorafenib is metabolised primarily in the liver and undergoes oxidative metabolism, mediated by CYP 3A4, as well as glucuronidation mediated by UGT1A9. Sorafenib conjugates may be cleaved in the gastrointestinal tract by bacterial glucuronidase activity, allowing reabsorption of unconjugated active substance. Co-administration of neomycin has been shown to interfere with this process, decreasing the mean bioavailability of sorafenib by 54%.
Sorafenib accounts for approximately 70-85% of the circulating analytes in plasma at steady state. Eight metabolites of sorafenib have been identified, of which five have been detected in plasma. The main circulating metabolite of sorafenib in plasma, the pyridine N-oxide, shows in vitro potency similar to that of sorafenib. This metabolite comprises approximately 9-16% of circulating analytes at steady state.
Following oral administration of a 100 mg dose of a solution formulation of sorafenib, 96% of the dose was recovered within 14 days, with 77% of the dose excreted in faeces, and 19% of the dose excreted in urine as glucuronidated metabolites. Unchanged sorafenib, accounting for 51% of the dose, was found in faeces but not in urine, indicating that biliary excretion of unchanged active substance might contribute to the elimination of sorafenib.
Pharmacokinetics in special populations: Analyses of demographic data suggest that there is no relationship between pharmacokinetics and age (up to 65 years), gender or body weight.
Paediatric population: No studies have been conducted to investigate the pharmacokinetics of sorafenib in paediatric patients.
Race: There are no clinically relevant differences in pharmacokinetics between Caucasian and Asian subjects.
Renal impairment: In four Phase I clinical trials, steady state exposure to sorafenib was similar in patients with mild or moderate renal impairment compared to the exposures in patients with normal renal function. In a clinical pharmacology study (single dose of 400 mg sorafenib), no relationship was observed between sorafenib exposure and renal function in subjects with normal renal function, mild, moderate or severe renal impairment. No data is available in patients requiring dialysis.
Hepatic impairment: In hepatocellular carcinoma (HCC) patients with Child-Pugh A or B (mild to moderate) hepatic impairment, exposure values were comparable and within the range observed in patients without hepatic impairment. The pharmacokinetics (PK) of sorafenib in Child-Pugh A and B non-HCC patients were similar to the PK in healthy volunteers. There are no data for patients with Child-Pugh C (severe) hepatic impairment. Sorafenib is mainly eliminated via the liver, and exposure might be increased in this patient population.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image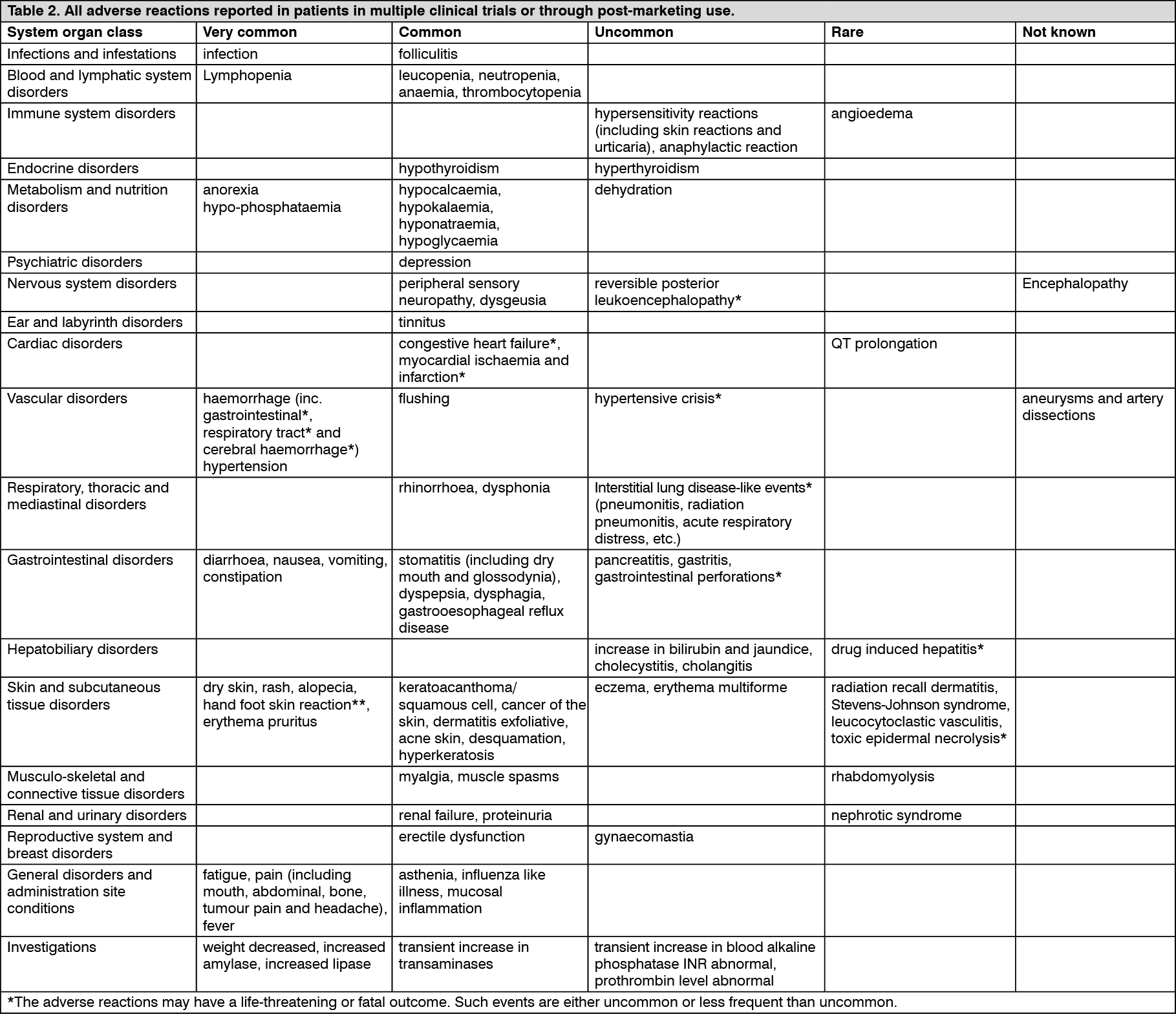 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
